- Research
JCAP researchers conducted Computational Investigation of single-atom bimetallic alloys
Proposed mechanism suggests that single-atom bimetallic alloys behave as a “one-pot” tandem catalysts, reducing CO2 to C1 hydrocarbon products.
Cheng, M.-J. et al. Quantum Mechanical Screening of Single-Atom Bimetallic Alloys for the Selective Reduction of CO2 to C1 Hydrocarbons. ACS Catalysis, DOI: 10.1021/acscatal.6b01393 (2016).
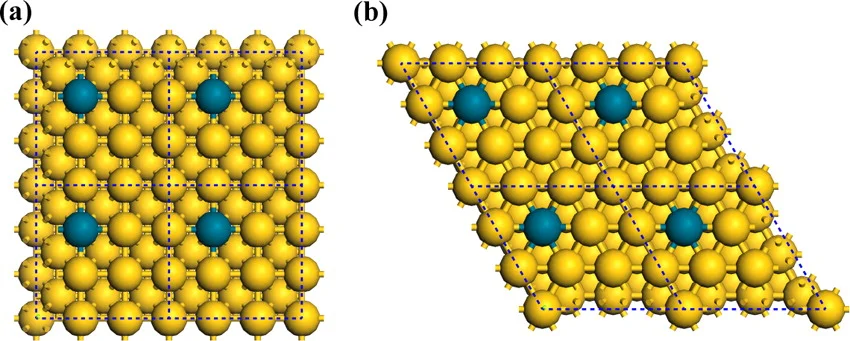
Reprinted with permission from Cheng, M.-J. et al. Quantum Mechanical Screening of Single-Atom Bimetallic Alloys for the Selective Reduction of CO2 to C1 Hydrocarbons. ACS Catalysis, DOI: 10.1021/acscatal.6b01393 (2016). Copyright ACS (2016).
Surface models used to simulate the single-atom alloys: (a) M@Au(111), M@Ag(111), and (b) M@Au(100), M@Ag(100). Each model is composed of a three-layer slab, using a 3 × 3 periodic cell, where one Au or Ag atom on the topmost layer is replaced by M (M = Cu, Ni, Pd, Pt, Co, Rh, and Ir). The periodic boundary is represented by the black dotted line.The most favorable kinetic pathways for the CO formation and formate formation pathway snapshots from AIMD simulations at 298 K and pH 7.
Understanding the mechanisms governing the reduction of carbon dioxide to hydrocarbons, which can be used as fuels, is a critical step towards development of new, efficient and selective catalytic materials. Quantum mechanical calculations have been used extensively to aid and further our knowledge of CO2 reduction reaction mechanisms. In the recent paper by Cheng et al., density functional theory (DFT) was used in combination with a Poisson-Boltzmann implicit solvation model to study the single-atom alloys (SAA) as electrocalaysts for CO2 reduction to C1 hydrocarbons. This computational approach offers an advantage of being computationally inexpensive so that relatively complex CO2 reduction reaction networks can be explored in depth.
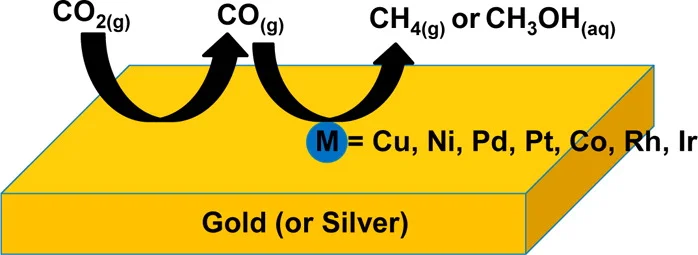
Reprinted with permission from Cheng, M.-J. et al. Quantum Mechanical Screening of Single-Atom Bimetallic Alloys for the Selective Reduction of CO2 to C1 Hydrocarbons. ACS Catalysis, DOI: 10.1021/acscatal.6b01393 (2016). Copyright ACS (2016).
Schematic Description of Our Proposed One-Pot Tandem Catalytic Reaction. The feedstock CO2 is first reduced to CO by gold or silver, and is subsequently captured and further reduced to C1 products by M.
The results provide a qualitative picture for the reaction that takes place on 28 different SAA consisting of well-defined silver (or gold) substrates in combination with isolated single metal (M) atoms (e.g., copper, nickel, rhodium, iridium, or platinum). The doping concentration in the top layer is approximately 11%.
The authors’ proposed mechanism suggests that SAAs behave as “one-pot” tandem catalysts, where silver (or gold) most efficiently reduce CO2 to CO, which forms a preferential bond with M to be further reduced to a hydrocarbon product. Out of the SAAs studied, 14 favored CO reduction over proton reduction, suggesting that those catalysts may preferentially suppress hydrogen generation in favor of CO2 reduction. Most of these materials contained cobalt, rhodium and iridium. Preferential binding of CO to M, also suggests that these SAAs are selective towards formation of C1 hydrocarbons.
This computational work offers encouraging motivation for experimental efforts to prepare and characterize SAAs for the electrochemical reduction of CO2.
This work is performed by the Joint Center for Artificial Photosynthesis, a DOE Energy Innovation Hub, supported through the Office of Science of the U.S. Department of Energy (Award No. DE-SC0004993).
Corresponding authors: alexbell@berkeley.edu and mhg@cchem.berkeley.edu