POSTER PRESENTATIONS
More than 100 researchers - graduate students, postdoctoral scholars, staff scientists and research engineers - work in JCAP laboratories at Caltech, LBNL, SLAC, Stanford, UC San Diego and Irvine. Their work is captured in posters presented here , covering a wide range of research fields, including materials discovery and characterization, developments of new experimental and theoretical methods, beam-line science, in-depth studies of catalytic mechanisms, and discovery of new solar-fuels generators.

We demonstrated high solar-to-H2 efficiency in PEC devices, consisting of a III-V based tandem light absorber and RuOx/Rh NP catalysts for OER and HER. Minimizing parasitic light absorption and reflection losses with favorable band alignment further reduces the efficiency gap to the theoretical limit. We
also developed a solar-driven CO2 reduction device using a gas diffusion electrode (GDE) with Ag nanoparticle catalyst directly powered by a III-V based
triple junction solar cell. Device geometry was studied to extend the operation stability.
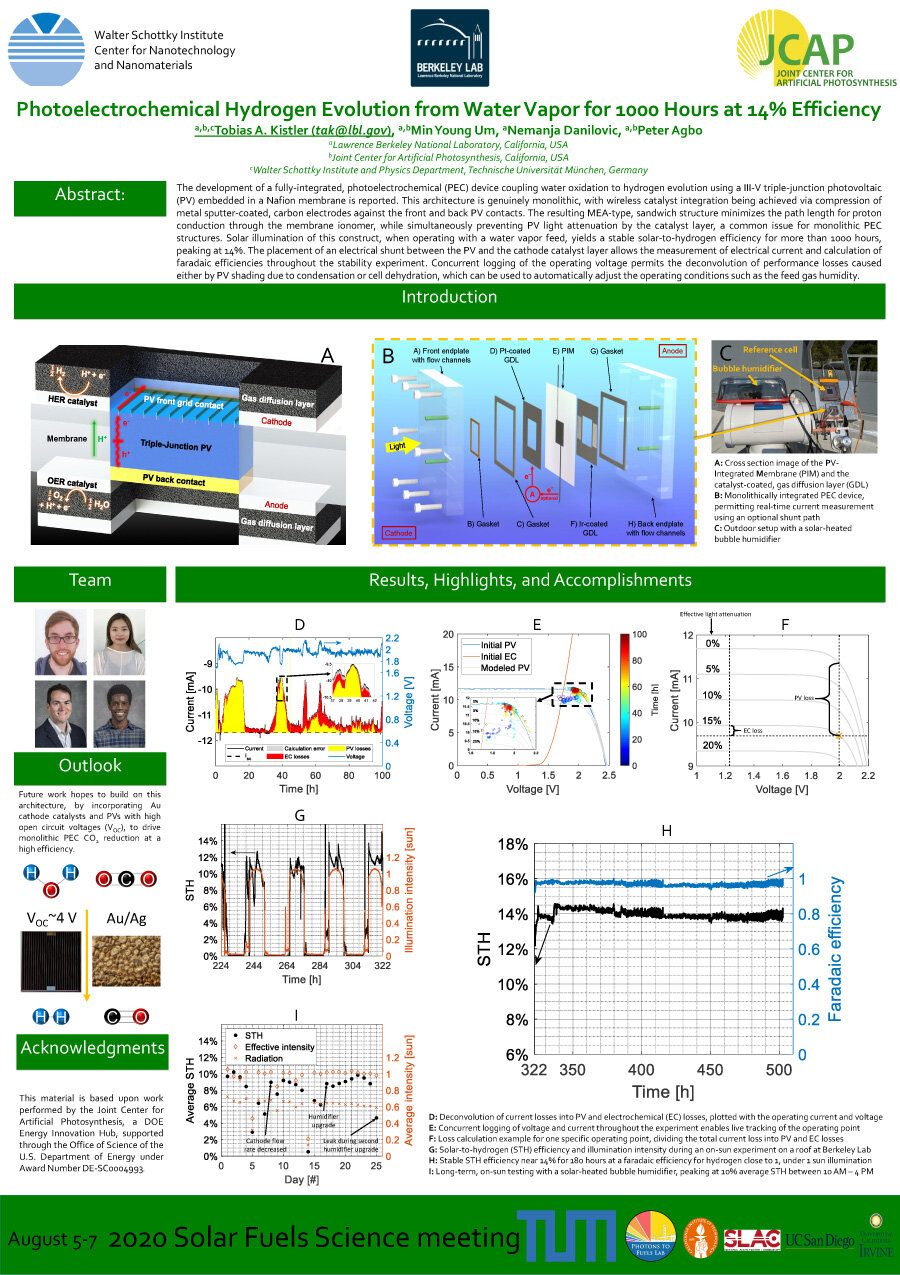
The development of a fully-integrated, photoelectrochemical (PEC) device coupling water oxidation to hydrogen evolution using a III-V triple-junction photovoltaic (PV) embedded in a Nafion membrane is reported. This architecture is genuinely monolithic, with wireless catalyst integration being achieved via compression of metal sputter-coated, carbon electrodes against the front and back PV contacts. The resulting MEA-type, sandwich structure minimizes the path length for proton conduction through the membrane ionomer, while simultaneously preventing PV light attenuation by the catalyst layer, a common issue for monolithic PEC structures. Solar illumination of this construct, when operating with a water vapor feed, yields a stable solar-to-hydrogen efficiency for more than 1000 hours, peaking at 14%. The placement of an electrical shunt between the PV and the cathode catalyst layer allows the measurement of electrical current and calculation of faradaic efficiencies throughout the stability experiment. Concurrent logging of the operating voltage permits the deconvolution of performance losses caused either by PV shading due to condensation or cell dehydration, which can be used to automatically adjust the operating conditions such as the feed gas humidity.

Designing a complete system to capture CO2 from the air and
transform it into valuable chemicals is of utmost importance for
mitigating climate change. The processes of CO2 sequestration, CO2 transformation, and product
separation all require significant energy inputs - therefore devising a system that simultaneously minimizes all of these steps is challenging. To date, a variety of CO2 sequestration and/or conversion systems have been built targeting these individual aspects. Here we propose a new paradigm for designing CO2 capture and conversion systems: (i) formation of bicarbonates/carbonates through dissolution of CO2 into basic solutions; followed by (ii) electrochemical reduction; and (iii) transformation into valuable chemicals via industrial processes. Unlike traditional systems in which gaseous CO2 reacts with a catalyst, our design focuses on the transformation of bicarbonate or carbonate ions from solution which offers several advantages. First, the CO2 sequestration from the atmosphere does not require an energy intensive heating step to recover gaseous CO2 for later transformation, and 75% of the CO2 is removed on the first pass. Second, by transforming bicarbonate/carbonate ions, the process avoids any energy intensive CO2 compression and allows for significantly higher conversion percentages. Taken together, we anticipate significant reductions in overall energy consumption can be achieved by focusing attention on the conversion of carbonate/bicarbonate ions instead of gaseous CO2.

In this poster, two approaches to light driven conversion of carbon dioxide to C2+ products are shown: (a) in a process analogous to natural photosynthesis, solar-driven reduction of carbon dioxide to hydrocarbon and oxygenate products is demonstrated with an overall efficiency exceeding 5%; and (b) solar-driven photocathode converts carbon dioxide to C 2 and C 3 products and has 20 day stability.
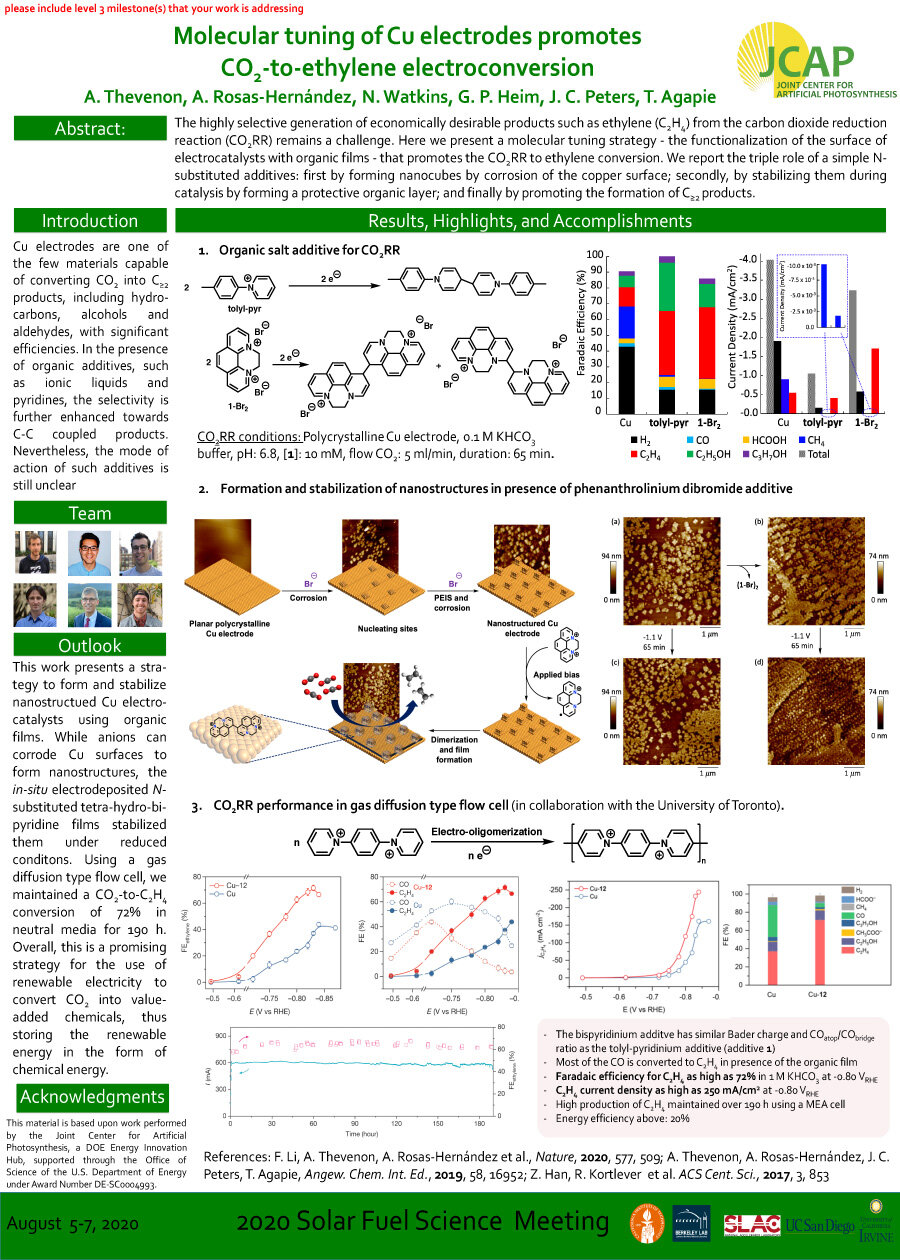
The highly selective generation of economically desirable products such as ethylene (C2H4) fromthe carbon dioxide reduction reaction (CO2RR) remains a challenge. Here we present a molecular tuning strategy - the functionalization of the surface of
electrocatalysts with organic films - that promotes the CO2RR to ethylene conversion.We report the triple role of a simple N-substituted additives: first by forming nanocubes by corrosion of the copper surface; secondly, by stabilizing them during catalysis by forming a protective organic layer; and finally by promoting the formation of C≥2 products.
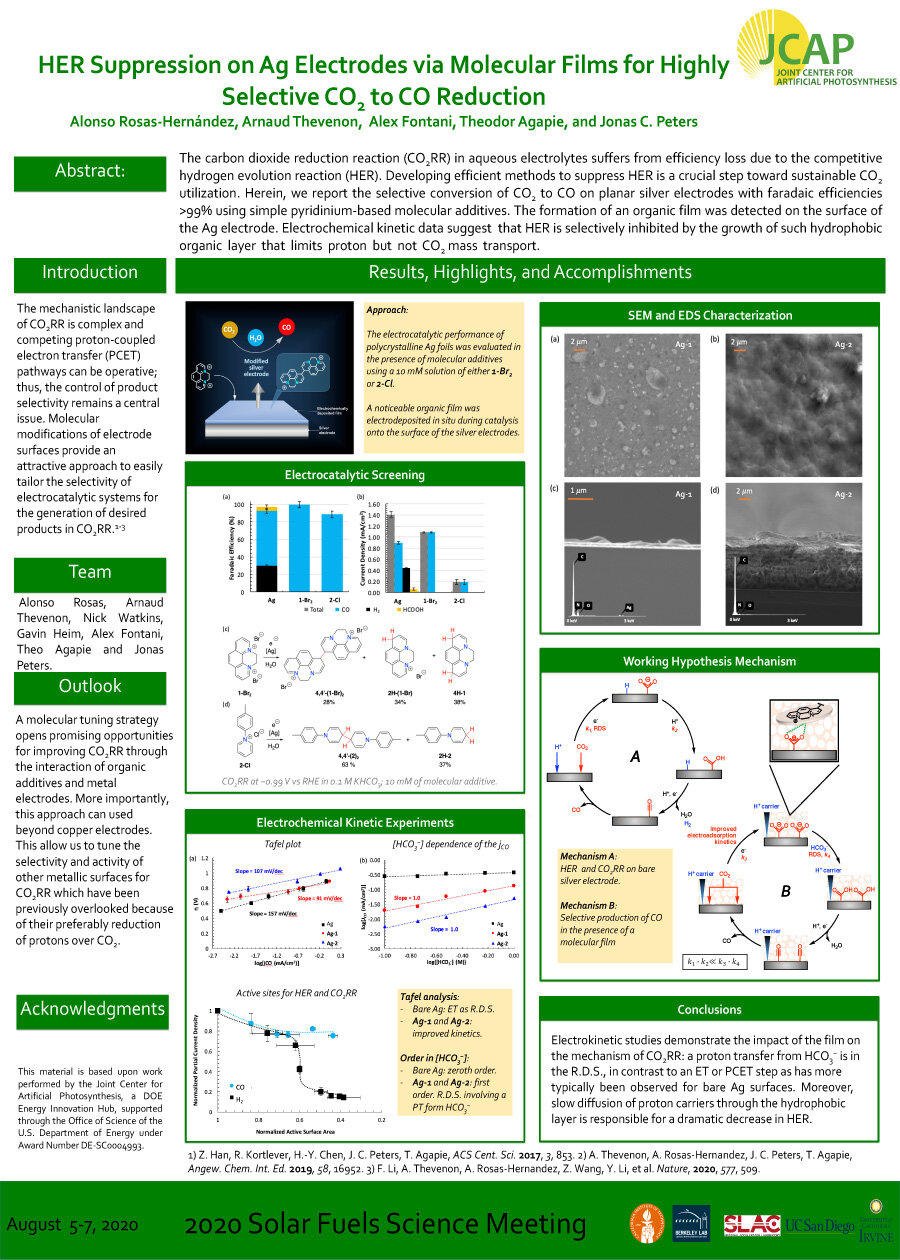
The carbon dioxide reduction reaction (CO2RR) in aqueous electrolytes suffers from efficiency loss due to the competitive hydrogen evolution reaction (HER). Developing efficient methods to suppress HER is a crucial step toward sustainable CO2 utilization. Herein, we report the selective conversion of CO2 to CO on planar silver electrodes with faradaic efficiencies
>99% using simple pyridinium-based molecular additives. The formation of an organic film was detected on the surface of the Ag electrode. Electrochemical kinetic data suggest that HER is selectively inhibited by the growth of such hydrophobic organic layer that limits proton but not CO2 mass transport.
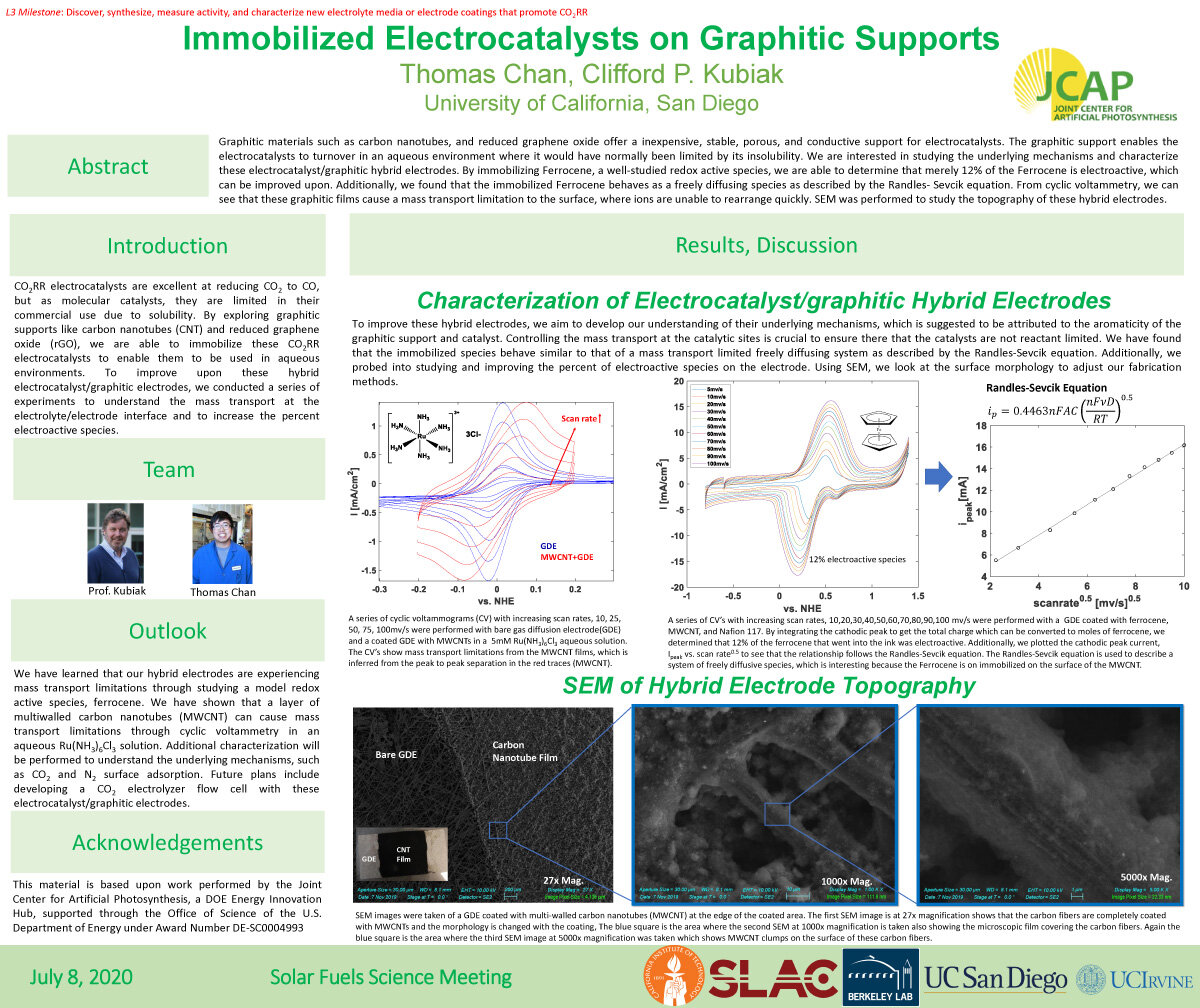
Graphitic materials such as carbon nanotubes, and reduced graphene oxide offer a inexpensive, stable, porous, and conductive support for electrocatalysts. The graphitic support enables the electrocatalysts to turnover in an aqueous environment where it would have normally been limited by its insolubility. We are interested in studying the underlying mechanisms and characterize these electrocatalyst/graphitic hybrid electrodes. By immobilizing Ferrocene, a well-studied redox active species, we are able to determine that merely 12% of the Ferrocene is electroactive, which
can be improved upon. Additionally, we found that the immobilized Ferrocene behaves as a freely diffusing species as described by the Randles- Sevcik equation. From cyclic voltammetry, we can see that these graphitic films cause a mass transport limitation to the surface, where ions are unable to rearrange quickly. SEM was performed to study the topography of these hybrid electrodes.

In the present study, we managed to propose and assess the practical pathways of electrochemical CO2RR that lead to the formation of C3 intermediates on the Cu(100) surface. We applied a combination of quantum mechanics, statistical mechanics, and electrodynamics, and evaluated the Gibbs free energies for all products, intermediates, and transition states. In the near future, we will complete the downstream reaction pathways that end in
the final C3 products[1] assuming negligible free energy barriers, and model the kinetics for the full reaction network.
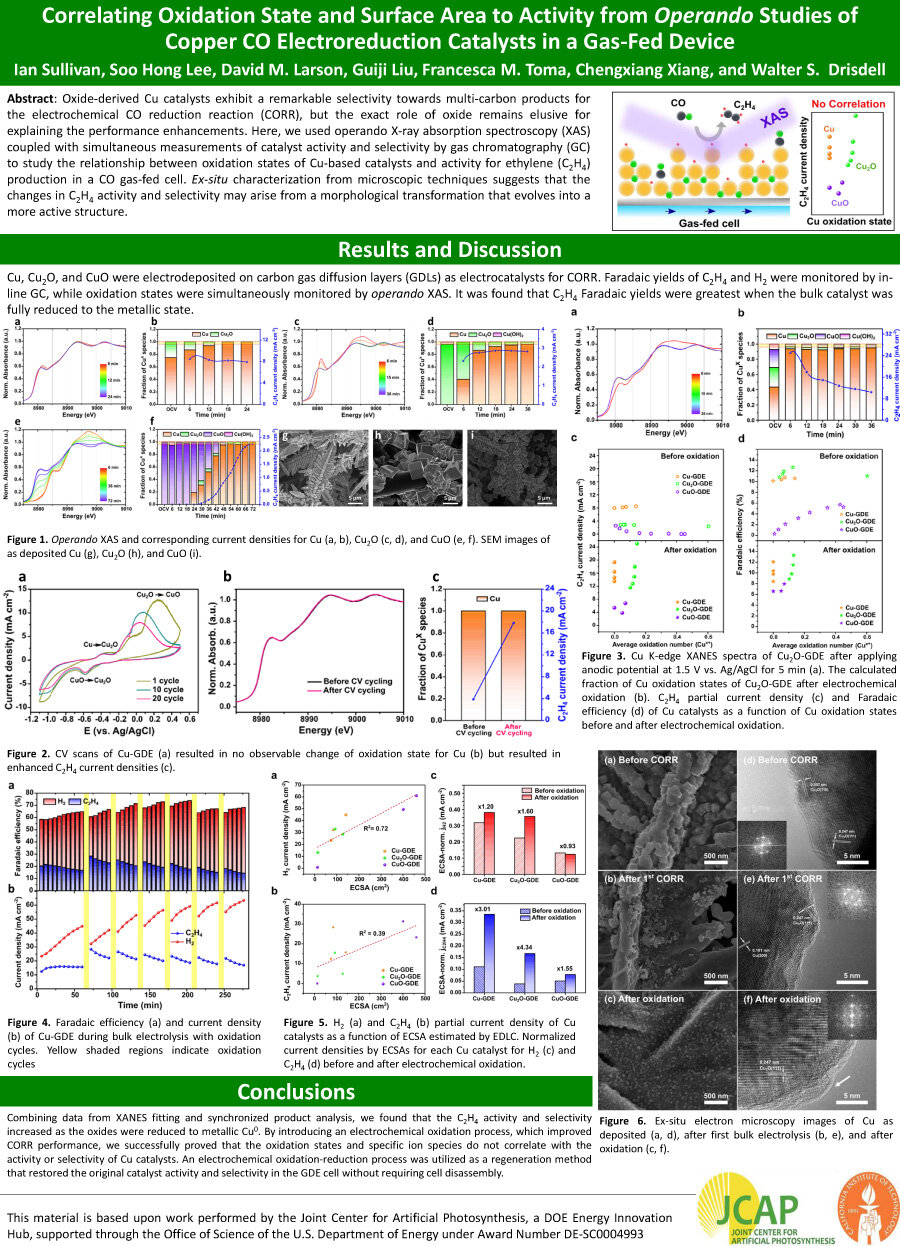
Oxide-derived Cu catalysts exhibit a remarkable selectivity towards multi-carbon products for
the electrochemical CO reduction reaction (CORR), but the exact role of oxide remains elusive for explaining the performance enhancements. Here, we used operando X-ray absorption spectroscopy (XAS)
coupled with simultaneous measurements of catalyst activity and selectivity by gas chromatography (GC)
to study the relationship between oxidation states of Cu-based catalysts and activity for ethylene (C2H4)
production in a CO gas-fed cell. Ex-situ characterization from microscopic techniques suggests that the changes in C2H4 activity and selectivity may arise from a morphological transformation that evolves into a
more active structure.
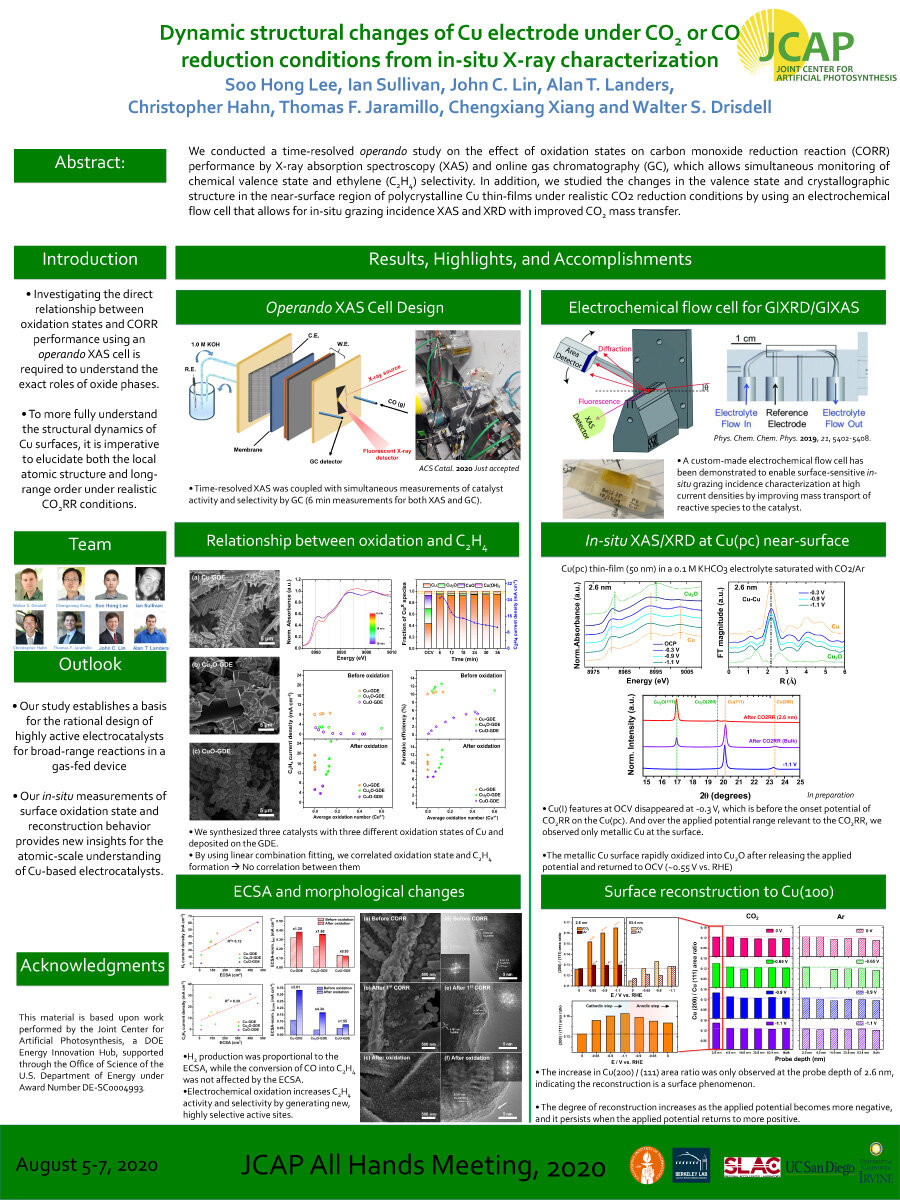
We conducted a time-resolved operando study on the effect of oxidation states on carbon monoxide reduction reaction (CORR) performance by X-ray absorption spectroscopy (XAS) and online gas chromatography (GC), which allows simultaneous monitoring of chemical valence state and ethylene (C2H4) selectivity. In addition, we studied the changes in the valence state and crystallographic structure in the near-surface region of polycrystalline Cu thin-films under realistic CO2 reduction conditions by using an electrochemical flow cell that allows for in-situ grazing incidence XAS and XRD with improvedCO2 mass transfer.
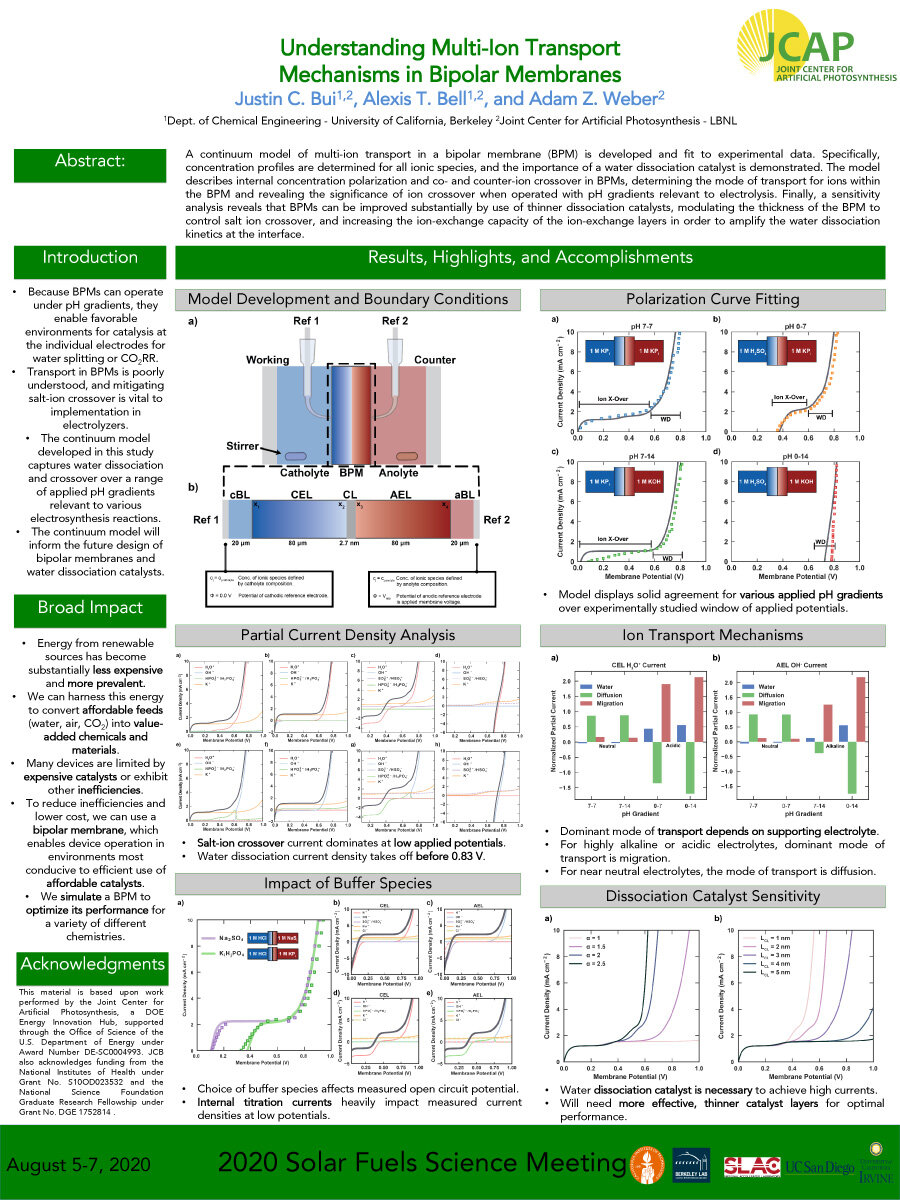
A continuum model of multi-ion transport in a bipolar membrane (BPM) is developed and fit to experimental data. Specifically, concentration profiles are determined for all ionic species, and the importance of a water dissociation catalyst is demonstrated. The model describes internal concentration polarization and co- and counter-ion crossover in BPMs, determining the mode of transport for ions within
the BPM and revealing the significance of ion crossover when operated with pH gradients relevant to electrolysis. Finally, a sensitivity analysis reveals that BPMs can be improved substantially by use of thinner dissociation catalysts, modulating the thickness of the BPM to control salt ion crossover, and increasing the ion-exchange capacity of the ion-exchange layers in order to amplify the water dissociation
kinetics at the interface.

Solar fuels generators operate under non-steady state conditions, however the permeability of the membranes used in them is represented using solubility and diffusivities obtained as fitting parameters from steady state measurements. Our work examines fundamental material properties and processes in order to build a model of membrane
permeation from physical chemistry that is realistic, valid at all times, and predictive for both steady state and non-steady state conditions. The multiscale reaction – diffusion glassy polymers, which examines the influence of the rigidity of the matrix when the solutes only interact weakly, and of methanol through Nafion, which examines a system that characterized by strong interactions. Permeation in all 3 systems is characterized by real-time polymeric matrix responses as the permeants are absorbed. This indicates that simple descriptions of membranes used in solar fuels generator models may not be sufficiently detailed to predict performance during the diurnal cycle, and that new studies of time-dependent polymer-solute interactions would be valuable.

In membrane electrode assemblies, the membrane plays an important role as it allows the transport of charge-carrier ions while inhibiting that of CO2 reduction products in order to prevent re-oxidation reactions which compromise the device’s overall efficiency. When running the device in a mildly alkaline environment with a highly negative working potential, the production of liquid fuel hydrocarbons increases while production of hydrogen decreases. However, as the working potential becomes increasingly negative,
device instability can also increase. The goal of this study is to develop an experimental approach that implements a highly negative working potential while maintaining a high and stable current density. Chronoamperometry (CA) was used to monitor device stability over time and potentiostatic electrochemical impedance spectroscopy (PEIS) was used to gauge the extent of membrane degradation.

The membrane is essential to maintaining efficiency within the photoelectrochemical cell because it mediates the transport of both CO2 reduction products and charge carriers. A better understanding of the physicochemical properties that mediate transport in these materials as they relate to performance in membrane-electrode assembly (MEA) type devices is needed. Herein, we modulated the
structure of anion-exchange membranes (AEMs) by controlling the synthesis of the polymer platform, which consists of a poly(phenylene oxide) (PPO) backbone and cationic imidazolium pendant groups. The changes in structure were correlated to the transport performance relevant to artificial photosynthesis devices. These studies revealed a tradeoff in the transport properties desirable for artificial photosynthesis: materials demonstrating high charger-carrier transport of also demonstrated high transport of reduction products.
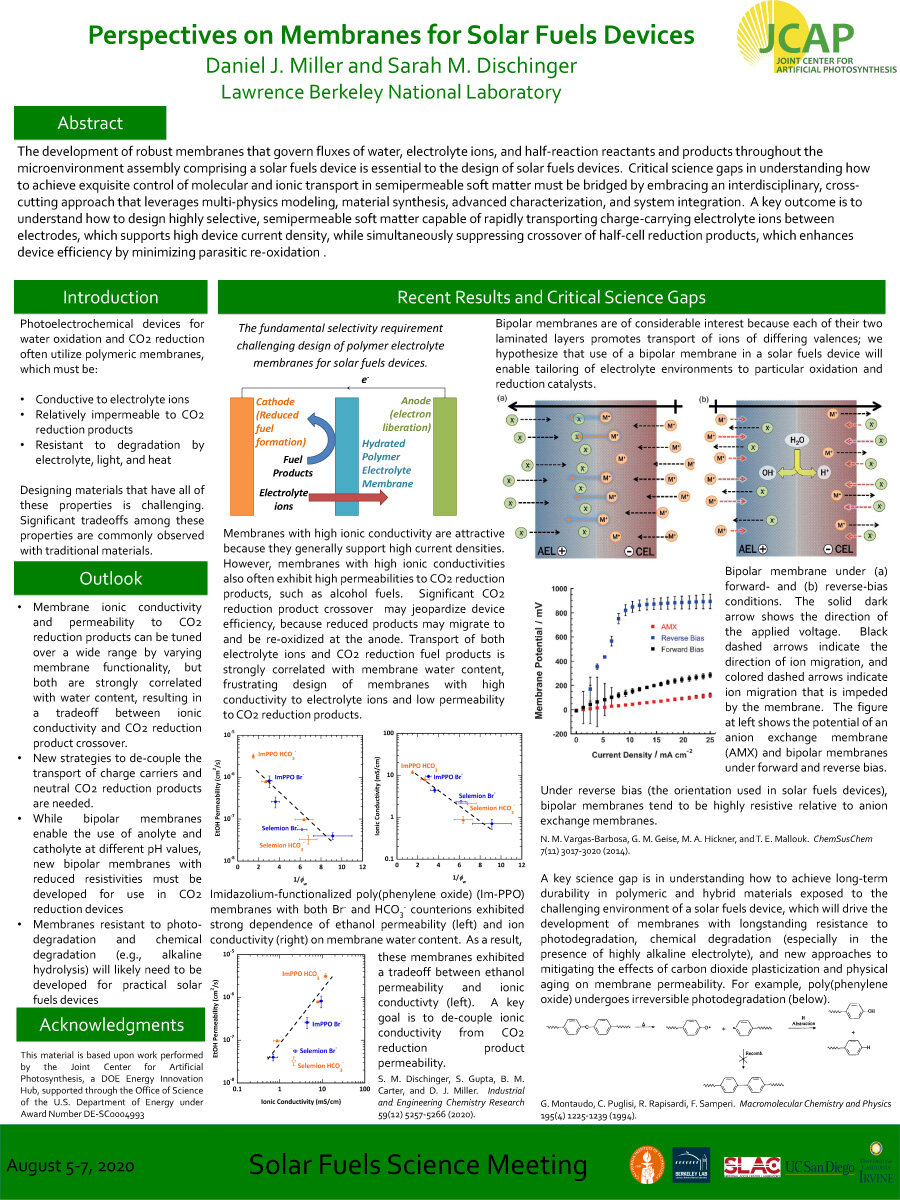
The development of robust membranes that govern fluxes of water, electrolyte ions, and half-reaction reactants and products throughout the microenvironment assembly comprising a solar fuels device is essential to the design of solar fuels devices. Critical science gaps in understanding how
to achieve exquisite control of molecular and ionic transport in semipermeable soft matter must be bridged by embracing an interdisciplinary, crosscutting approach that leverages multi-physics modeling, material synthesis, advanced characterization, and system integration. A key outcome is to understand how to design highly selective, semipermeable soft matter capable of rapidly transporting charge-carrying electrolyte ions between electrodes, which supports high device current density, while simultaneously suppressing crossover of half-cell reduction products, which enhances device efficiency by minimizing parasitic re-oxidation.
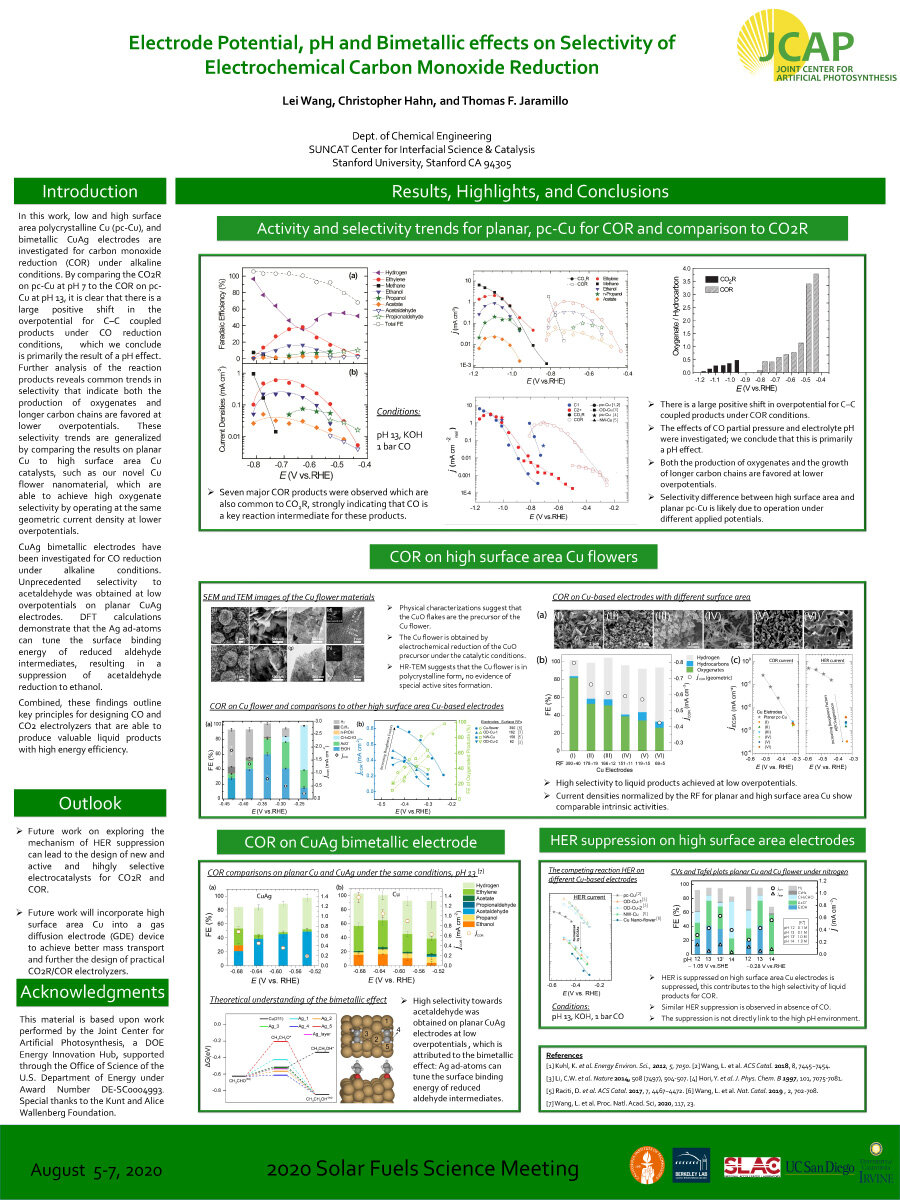
In this work, low and high surface area polycrystalline Cu (pc-Cu), and bimetallic CuAg electrodes are investigated for carbon monoxide reduction (COR) under alkaline conditions. By comparing the CO2R on pc-Cu at pH 7 to the COR on pc- Cu at pH 13, it is clear that there is a large positive shift in the overpotential for C–C coupled products under CO reduction conditions, which we conclude is primarily the result of a pH effect. Further analysis of the reaction products reveals common trends in selectivity that indicate both the production of oxygenates and longer carbon chains are favored at lower overpotentials. These selectivity trends are generalized by comparing the results on planar Cu to high surface area Cu catalysts, such as our novel Cu flower nanomaterial, which are able to achieve high oxygenate selectivity by operating at the same geometric current density at lower overpotentials.

The development of efficient, stable photoelectrodes remain a primary materials challenge for solar fuels generation. The photoanode is needed to provide protons and electrons to the (photo)cathode, while development of a CO2RR-active photocathode provides opportunities to steer product selectivity, with both photoelectrodes providing energy gain for fuel formation. We demonstrate efficient high throughput evaluation of the performance of photoelectrode assemblies consisting of compositionally diverse metal oxide coatings on light absorbers, which reveals the critical role of effective surface passivation, and the inter-connected performance impacts of coating composition and loading, and electrolyte pH.

A screening procedure using density functional theory (DFT) was devised to select promising materials for the CO2 reduction reaction (CO2RR) from the Materials Project (MP) database. Criteria such as aqueous and thermodynamic stability, computational cost, and photochemical suitability were considered. The selected materials are further investigated using a high-throughput workflow for generating adsorption data for semiconductor surfaces. By considering species relevant to the CO2RR as adsorbates, the descriptors computed by the workflow offer additional insight into the photocatalytic performance of the researched materials, making this approach a powerful materials discovery tool.

Peterson et al.
First-principles ab initio calculations are a powerful tool for understanding the electronic structure of photo(electro)catalyst materials for artificial photosynthesis. In collaboration with the Gregoire group we study the novel high-photovoltage photoanode n-type FeWO4 including its electronic structure and its hypothesized synthesis process. In collaboration with the Atwater group we also study MoS2(1-x)Se2x alloys for photo(electro)cathode applications, including a discussion of different crystal structure morphologies and their respective band
edge alignment to CO2 reduction reaction potentials.
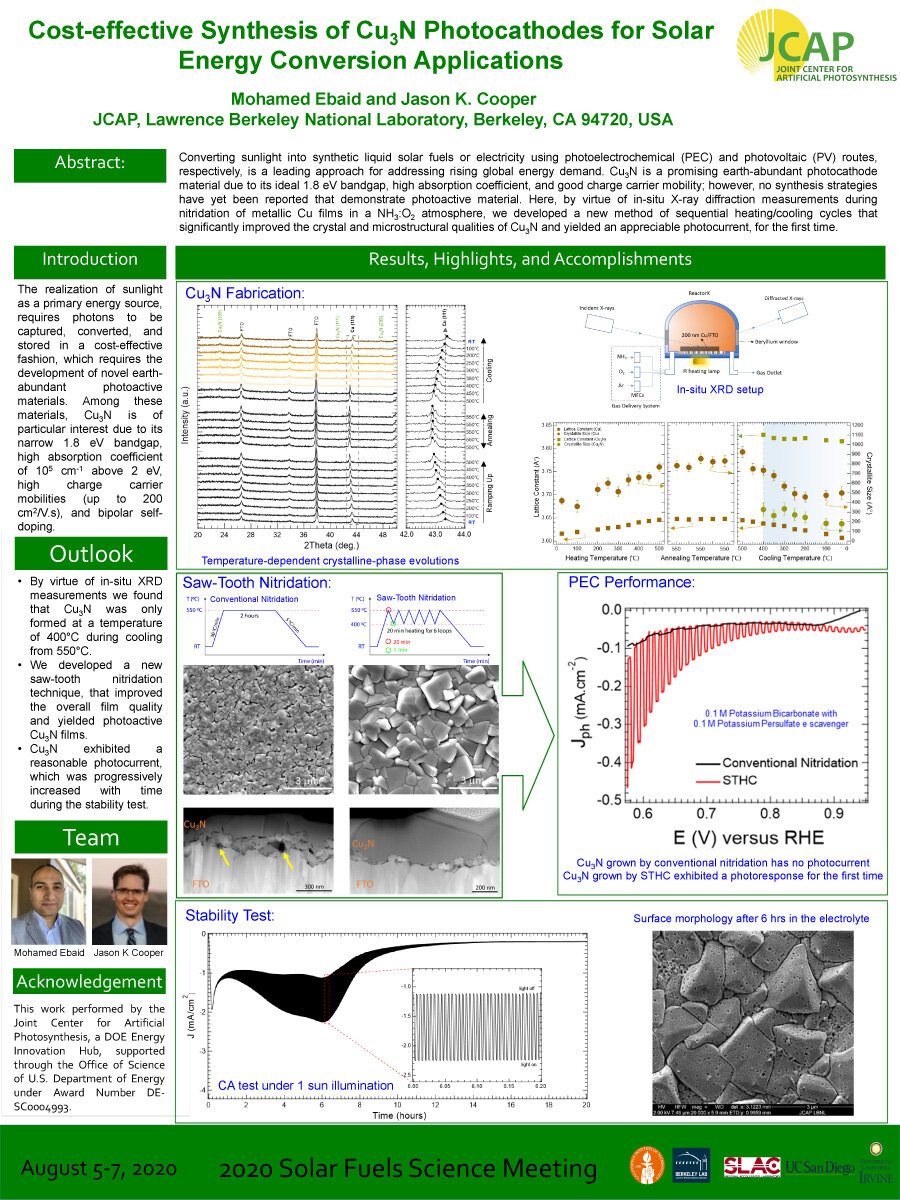
Converting sunlight into synthetic liquid solar fuels or electricity using photoelectrochemical (PEC) and photovoltaic (PV) routes, respectively, is a leading approach for addressing rising global energy demand. Cu3N is a promising earth-abundant photocathode
material due to its ideal 1.8 eV bandgap, high absorption coefficient, and good charge carrier mobility; however, no synthesis strategies have yet been reported that demonstrate photoactive material. Here, by virtue of in-situ X-ray diffraction measurements during nitridation of metallic Cu films in a NH3:O2 atmosphere, we developed a new method of sequential heating/cooling cycles that significantly improved the crystal and microstructural qualities of Cu3N and yielded an appreciable photocurrent, for the first time.

Electrocatalysis – the action of a heterogeneous catalyst under electrochemical conditions – is a surface-mediated process. Activity and selectivity are dependent upon the composition and the crystallographic structure of the electrode surface, the nature of the reactant-catalyst interactions, and the applied electrode potential. The surface science approach to electrocatalysis entails the scrutiny of surfaces with well-defined structure and composition before, during, and after the reaction of interest so that unambiguous correlations can be drawn. The merits of the often slow and invariably rigorous electrochemical surface science protocols are showcased herein for CO2 reduction studies. Systematic investigations are built upon copper, the sole monometallic electrocatalyst for the production of a variety of energy-rich molecules. The development and the seriatim implementation of operando tools have led to the discovery of (i) the potential-driven surface reconstruction of polycrystalline Cu into a (100)-terminated surface; (ii) the relative stability of the low-index facets of Cu; (iii) the atomic details of the chemisorption of CO on Cu(100) under different electrolytic environments; and (iv) the specific surface structure selective for the formation of ethanol, C2H5OH, in alkaline medium.
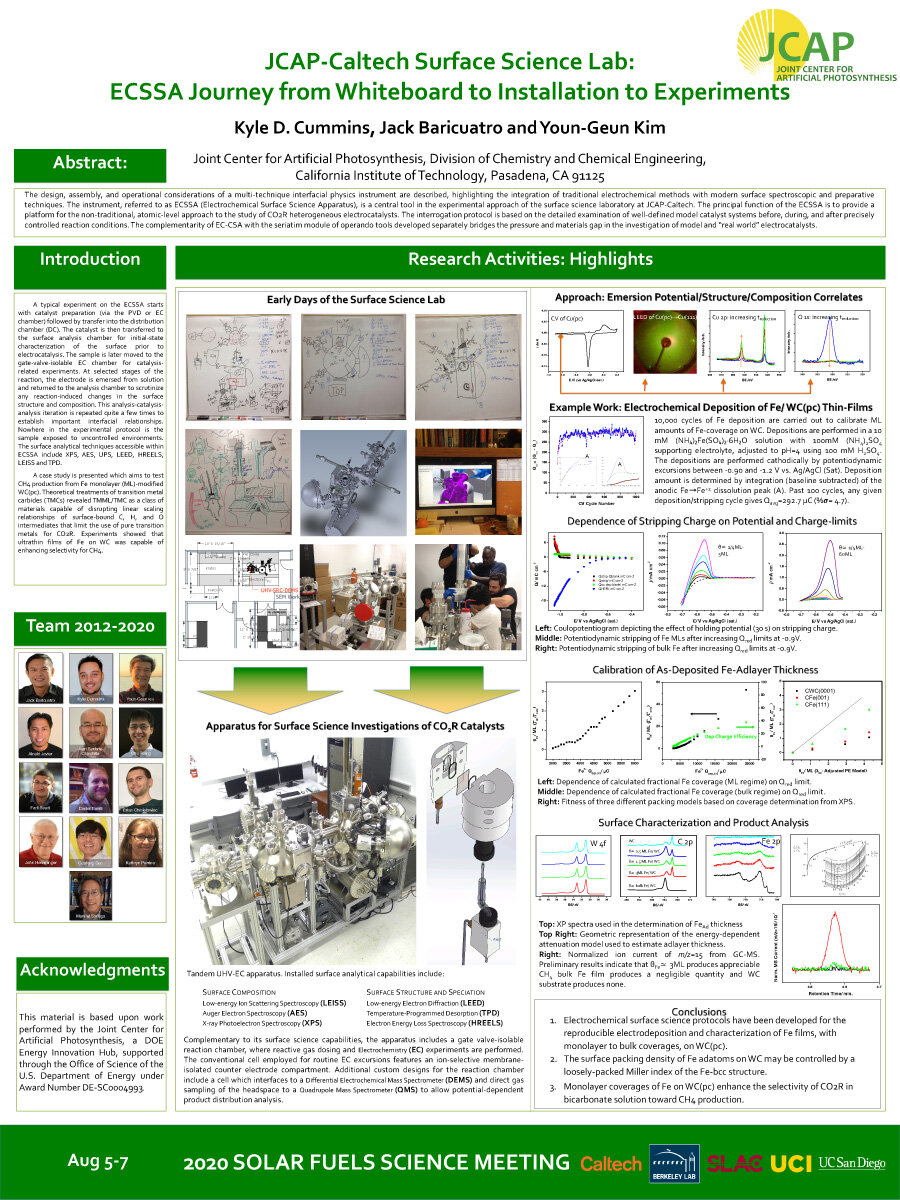
The design, assembly, and operational considerations of a multi-technique interfacial physics instrument are described, highlighting the integration of traditional electrochemical methods with modern surface spectroscopic and preparative techniques. The instrument, referred to as ECSSA (Electrochemical Surface Science Apparatus), is a central tool in the experimental approach of the surface science laboratory at JCAP-Caltech. The principal function of the ECSSA is to provide a platform for the non-traditional, atomic-level approach to the study of CO2R heterogeneous electrocatalysts. The interrogation protocol is based on the detailed examination of well-defined model catalyst systems before, during, and after precisely controlled reaction conditions.The complementarity of EC-CSA with the seriatim module of operando tools developed separately bridges the pressure and materials gap in the investigation of model and “real world” electrocatalysts.
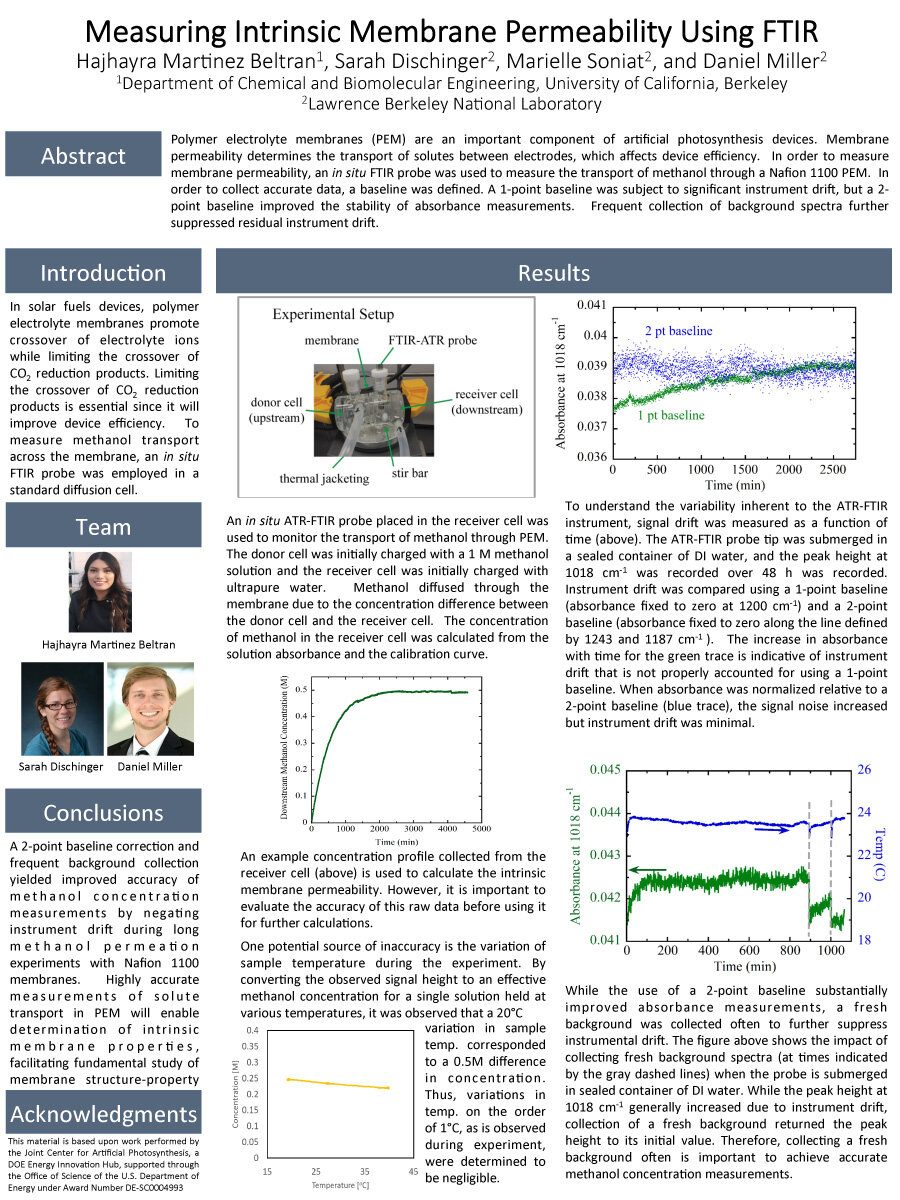
Polymer electrolyte membranes (PEM) are an important component of artificial photosynthesis devices. Membrane permeability determines the transport of solutes between electrodes, which affects
device efficiency. In order to measure membrane permeability, an in situ FTIR probe was used to measure the transport of methanol through a Nafion 1100 PEM. In order to collect accurate data, a baseline was defined. A 1-point baseline was subject to significant instrument drift, but a 2-point baseline
improved the stability of absorbance measurements. Frequent collection of background spectra further suppressed residual instrument drift.
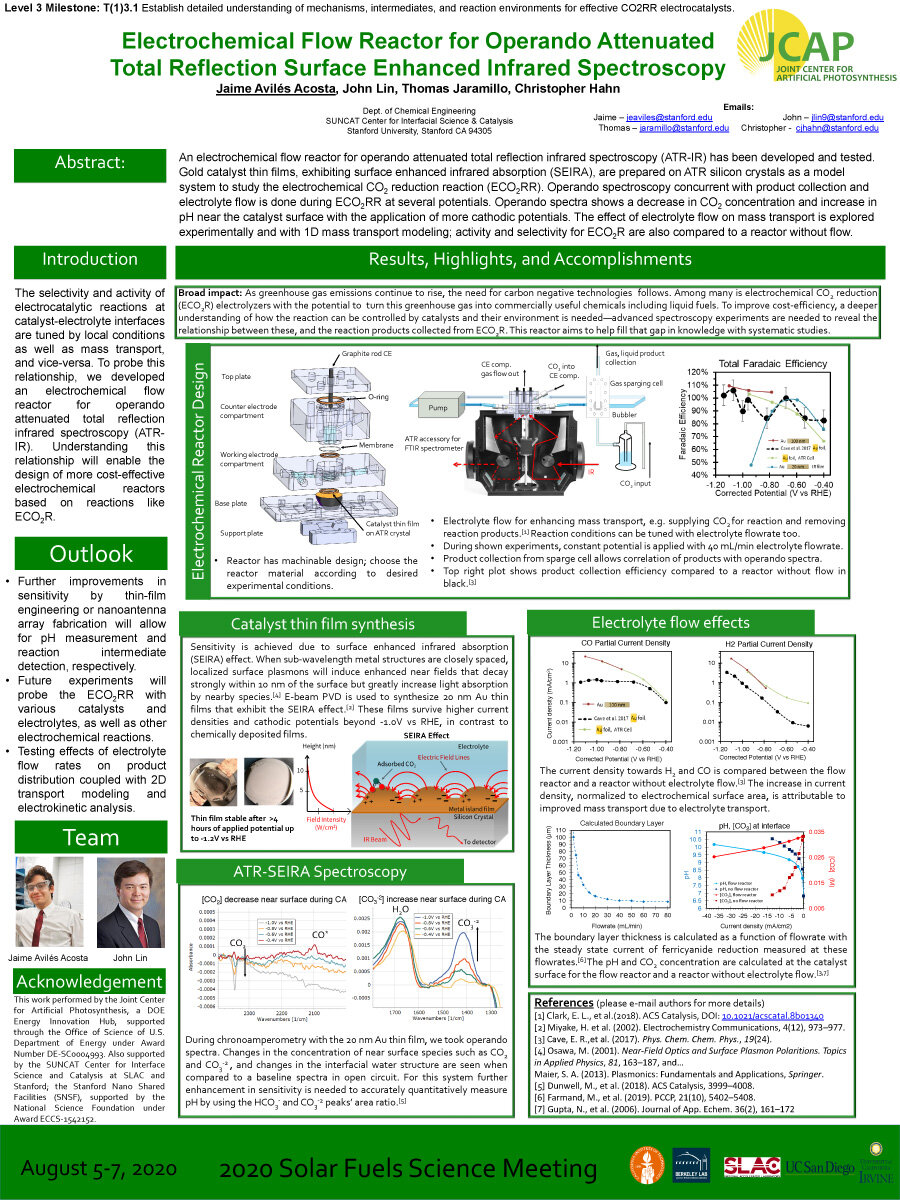
An electrochemical flow reactor for operando attenuated total reflection infrared spectroscopy (ATR-IR) has been developed and tested. Gold catalyst thin films, exhibiting surface enhanced infrared absorption (SEIRA), are prepared on ATR silicon crystals as a model system to study the electrochemical CO2 reduction reaction (ECO2RR). Operando spectroscopy concurrent with product collection and electrolyte flow is done during ECO2RR at several potentials. Operando spectra shows a decrease in CO2 concentration and increase in pH near the catalyst surface with the application of more cathodic potentials. The effect of electrolyte flow on mass transport is explored experimentally and with 1D mass transport modeling; activity and selectivity for ECO2R are also compared to a reactor without flow.
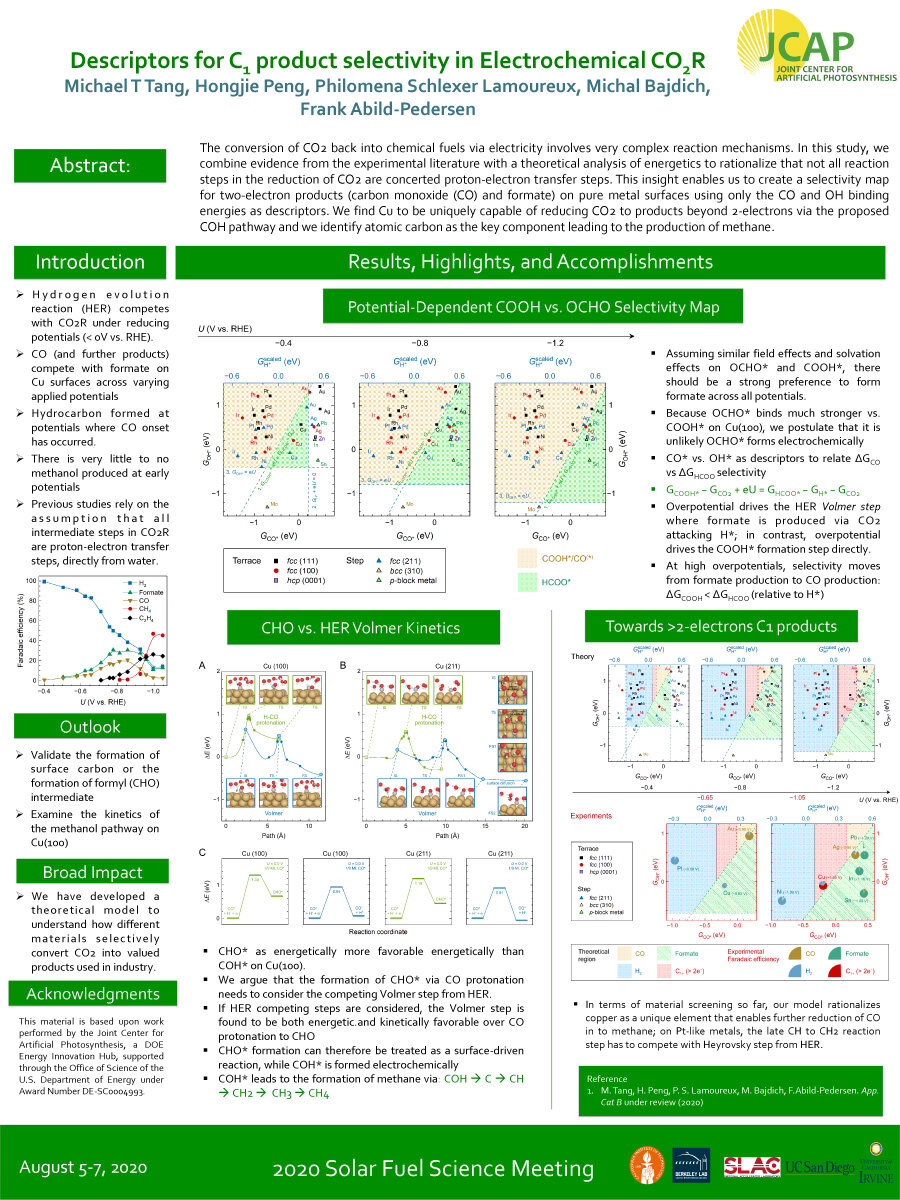
The conversion of CO2 back into chemical fuels via electricity involves very complex reaction mechanisms. In this study, we combine evidence from the experimental literature with a theoretical analysis of energetics to rationalize that not all reaction
steps in the reduction of CO2 are concerted proton-electron transfer steps. This insight enables us to create a selectivity map for two-electron products (carbon monoxide (CO) and formate) on pure metal surfaces using only the CO and OH binding energies as descriptors. We find Cu to be uniquely capable of reducing CO2 to products beyond 2-electrons via the proposed COH pathway and we identify atomic carbon as the key component leading to the production of methane.

The lack of first-principle's methods to directly reveal the processes of carrier transfers makes it challenging to understand the fundamentals of electrochemical systems. By using the recently developed non-adiabatic molecular dynamics (NA-MD) and real-time time-dependent density functional theory (rt-TDDFT), we illustrate the full profiles of hot carrier cooling in interfacial systems and explore the significance of non-adiabaticity (NA) in reactions. The Schottky barriers and device design strategy could strongly suppress the back-transfer and enhance charge separation. By using one step of CO2 reaction as an example, we find that conventional ground-state methods calculated reaction barriers could be underestimated. Moreover, we are developing new methodologies to expand current NA-MD and TDDFT capability. By including many-body effects, exciton dynamics in low-dimensional materials can be studied by first-principle. Molecular damage under solvent is also explored with wavefunction collapsing method.
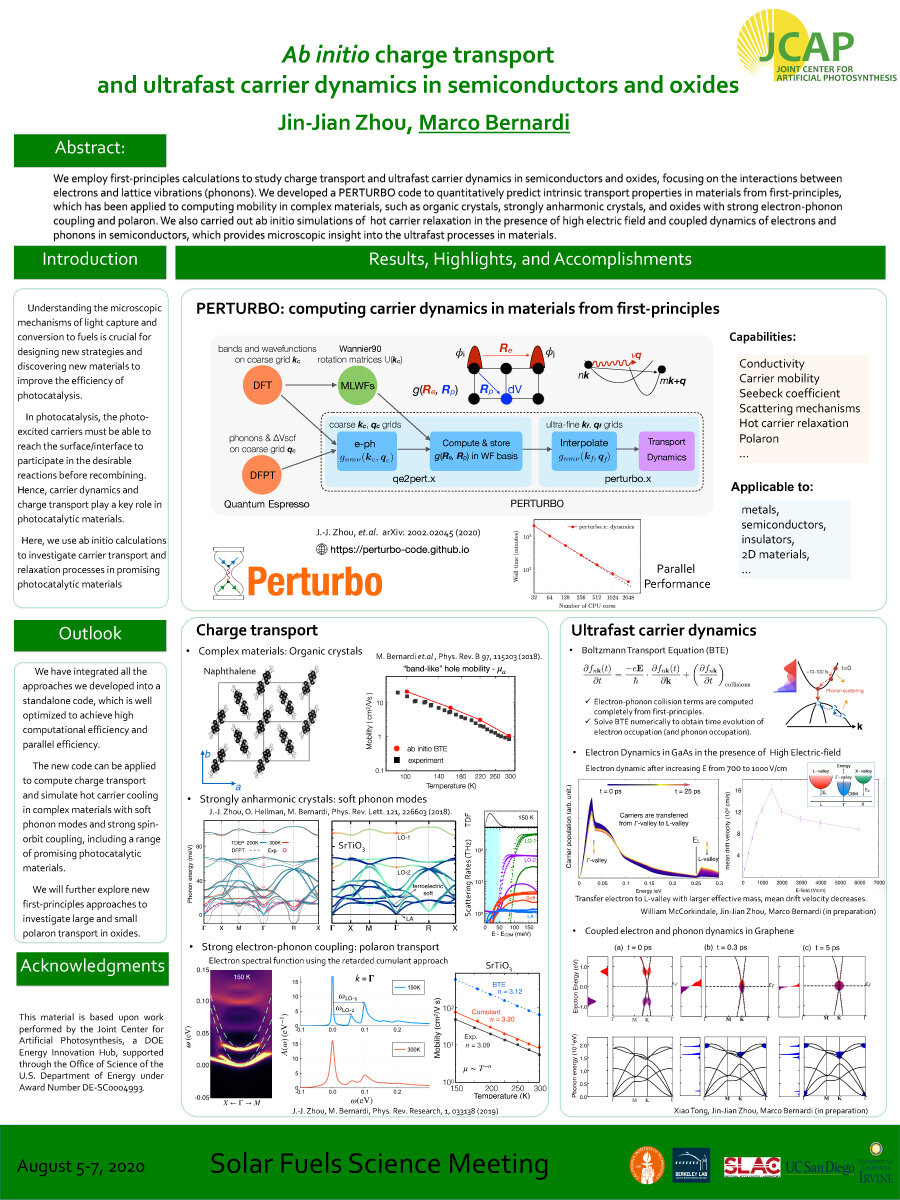
We employ first-principles calculations to study charge transport and ultrafast carrier dynamics in semiconductors and oxides, focusing on the interactions between electrons and lattice vibrations (phonons). We developed a PERTURBO code to quantitatively predict intrinsic transport properties in materials from first-principles, which has been applied to computing mobility in complex materials, such as organic crystals, strongly anharmonic crystals, and oxides with strong electron-phonon coupling and polaron. We also carried out ab initio simulations of hot carrier relaxation in the presence of high electric field and coupled dynamics of electrons and phonons in semiconductors, which provides microscopic insight into the ultrafast processes in materials.
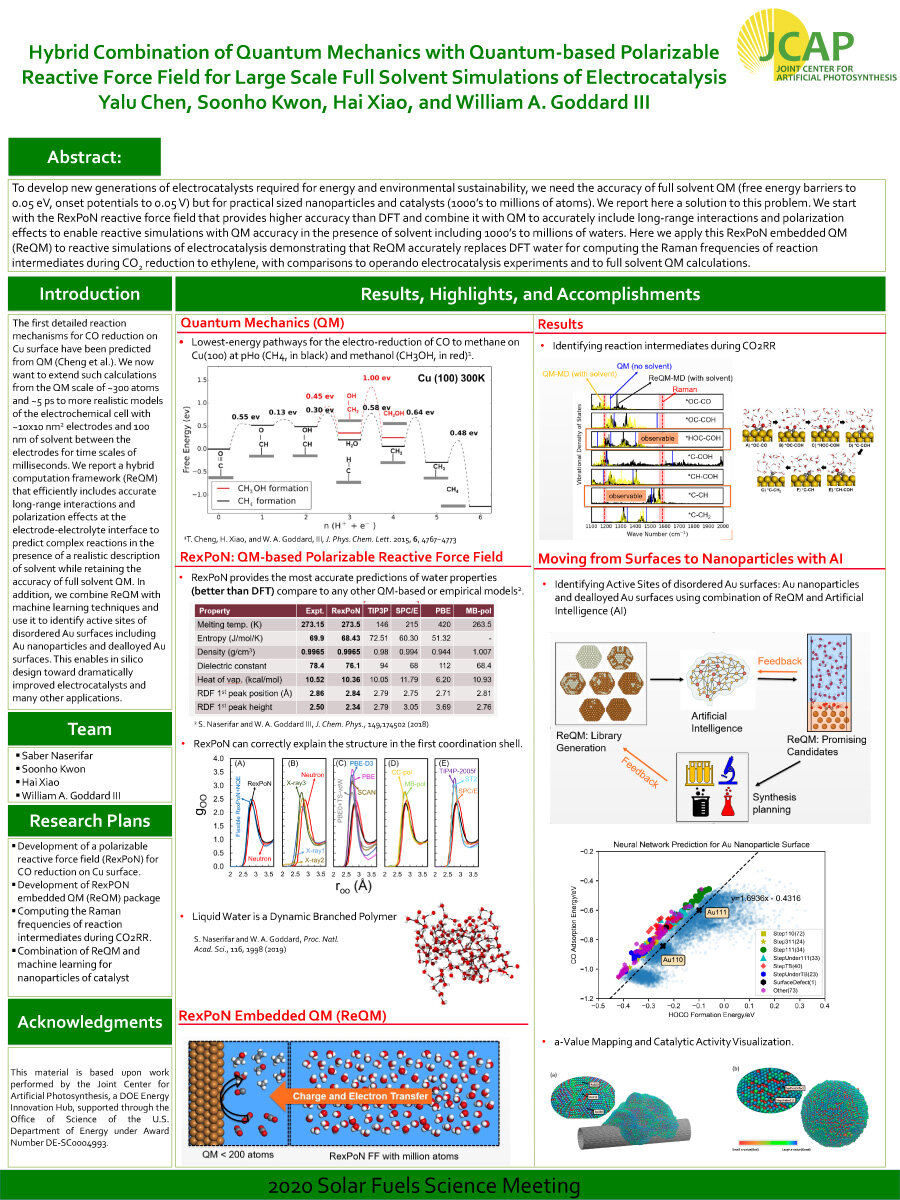
To develop new generations of electrocatalysts required for energy and environmental sustainability, we need the accuracy of full solvent QM (free energy barriers to 0.05 eV, onset potentials to 0.05 V) but for practical sized nanoparticles and catalysts (1000’s to millions of atoms). We report here a solution to this problem. We start with the RexPoN reactive force field that provides higher accuracy than DFT and combine it with QM to accurately include long-range interactions and polarization effects to enable reactive simulations with QM accuracy in the presence of solvent including 1000’s to millions of waters. Here we apply this RexPoN embedded QM (ReQM) to reactive simulations of electrocatalysis demonstrating that ReQM accurately replaces DFT water for computing the Raman frequencies of reaction intermediates during CO2 reduction to ethylene, with comparisons to operando electrocatalysis experiments and to full solvent QM calculations.
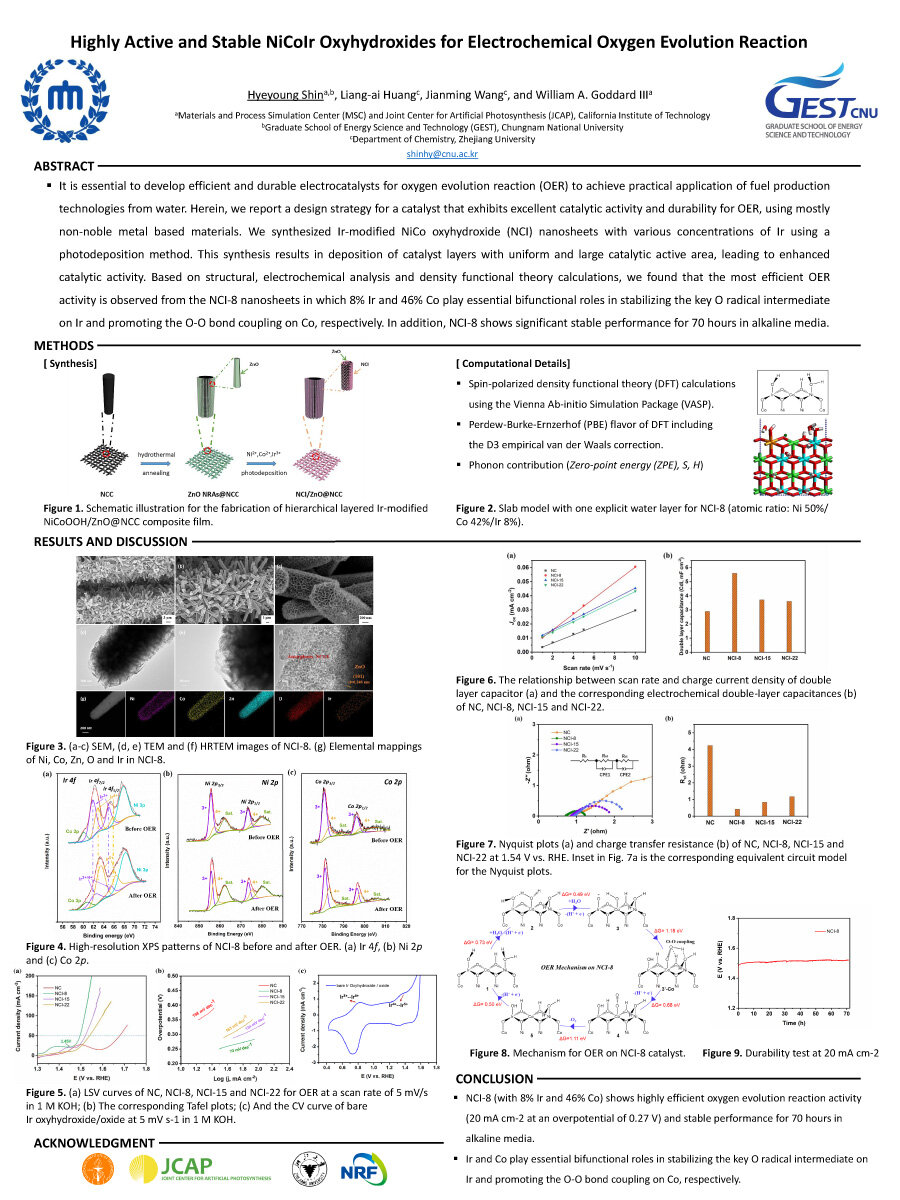
It is essential to develop efficient and durable electrocatalysts for oxygen evolution reaction (OER) to achieve practical application of fuel production technologies from water. Herein, we report a design strategy for a catalyst that exhibits excellent catalytic activity and durability for OER, using mostly non-noble metal based materials. We synthesized Ir-modified NiCo oxyhydroxide (NCI) nanosheets with various concentrations of Ir using a photodeposition method. This synthesis results in deposition of catalyst layers with uniform and large catalytic active area, leading to enhanced catalytic activity. Based on structural, electrochemical analysis and density functional theory calculations, we found that the most efficient OER activity is observed from the NCI-8 nanosheets in which 8% Ir and 46% Co play essential bifunctional roles in stabilizing the key O radical intermediate
on Ir and promoting the O-O bond coupling on Co, respectively. In addition, NCI-8 shows significant stable performance for 70 hours in alkaline media.

Mixed halide perovskites sparked great research interest due to their outstanding optoelectronic properties, ease of fabrication and bandgap tunability. Within the ABX3 structure, especially the composition of the X-site is varied to tune the bandgap, mostly using iodide and bromide. Those mixed perovskites however suffer from phase instabilities under
illumination in which microscopic clusters with high iodide content are formed and act as recombination centers. The key mechanism(s) underlying this halide segregation process are still debated. We investigated the influence of microstructure and in particular, grain size and heterogeneity, on this process for the archetype perovskite MAPb(I1.5,Br1.5). Our findings show that the segregation process occurs in three stages (not two as commonly reported) and starts with a flash formation of I-rich nano domains. The composition of those nano domains depends on the grain size due to the prevalent compositional fluctuations as well as the abundance of defect states at grain boundaries.
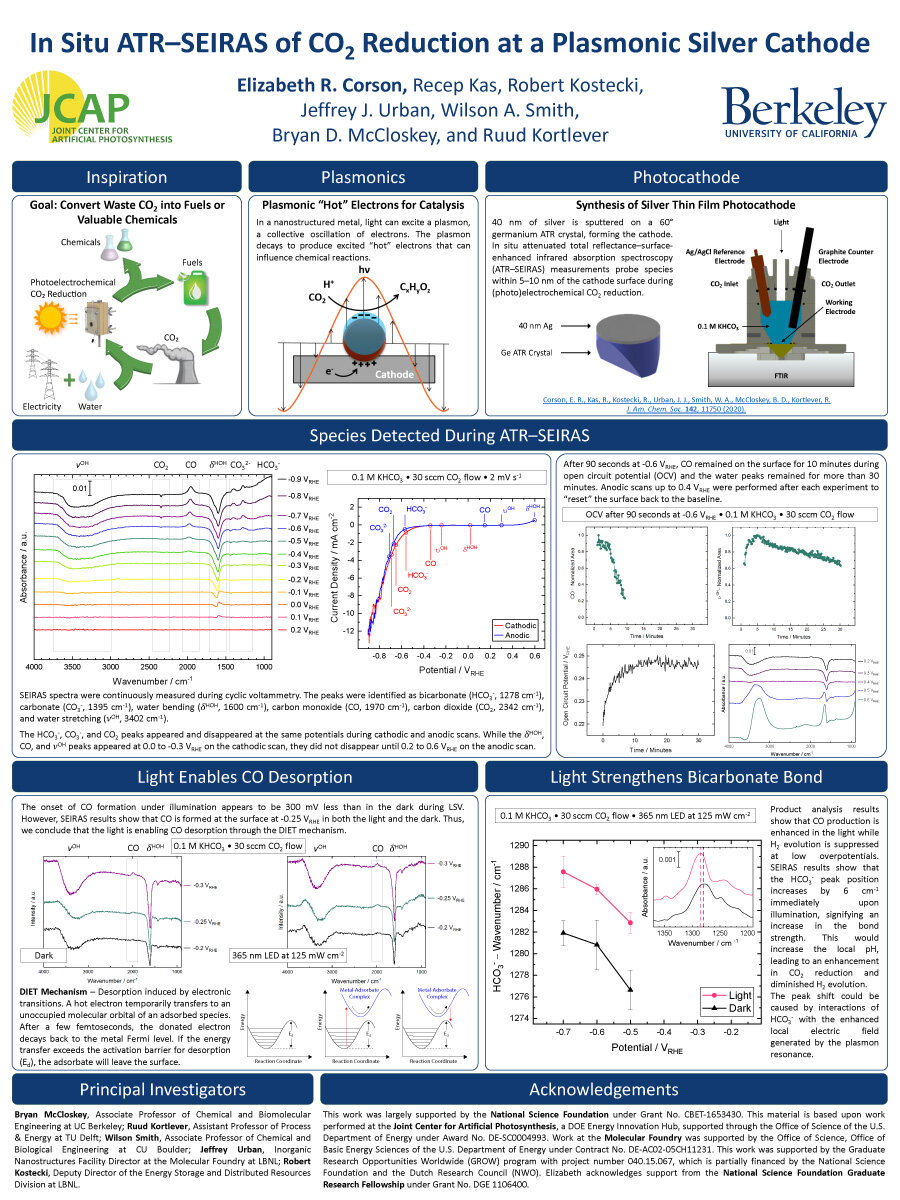
Illumination of a voltage-biased plasmonic silver cathode during carbon dioxide reduction results in a suppression of the hydrogen evolution reaction while enhancing carbon dioxide reduction. This effect has been shown to be photonic rather than thermal, but the exact plasmonic mechanism is unknown. Here, we conduct an in situ ATR−SEIRAS (attenuated total reflectance−surface-enhanced infrared absorption spectroscopy) study of a sputtered thin film silver cathode on a germanium ATR crystal in carbon dioxide-saturated 0.1 M potassium bicarbonate over a range of potentials under both dark and illuminated conditions to elucidate the nature of this plasmonic enhancement.
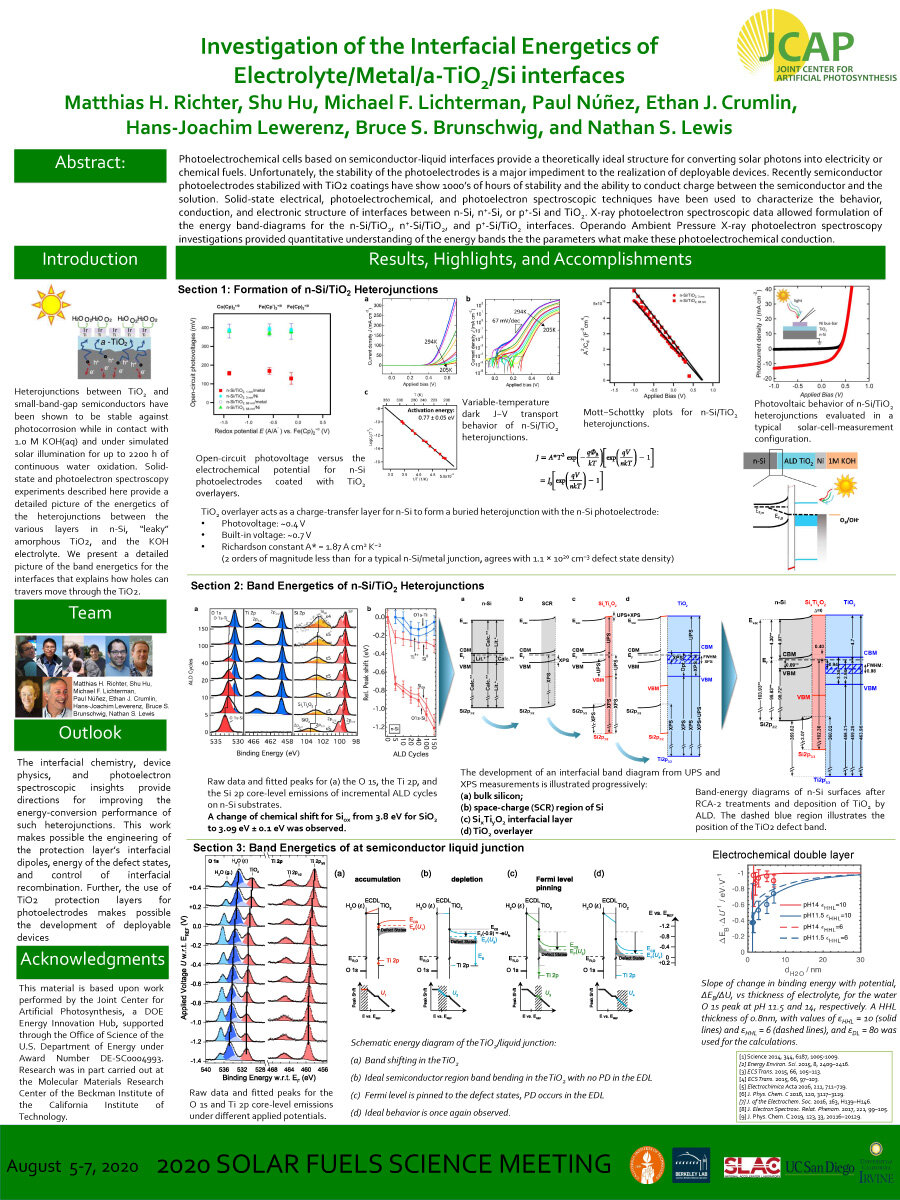
Photoelectrochemical cells based on semiconductor-liquid interfaces provide a theoretically ideal structure for converting solar photons into electricity or
chemical fuels. Unfortunately, the stability of the photoelectrodes is a major impediment to the realization of deployable devices. Recently semiconductor photoelectrodes stabilized with TiO2 coatings have show 1000’s of hours of stability and the ability to conduct charge between the semiconductor and the solution. Solid-state electrical, photoelectrochemical, and photoelectron spectroscopic techniques have been used to characterize the behavior, conduction, and electronic structure of interfaces between n-Si, n+-Si, or p+-Si and TiO2. X-ray photoelectron spectroscopic data allowed formulation of the energy band-diagrams for the n-Si/TiO2, n+-Si/TiO2, and p+-Si/TiO2 interfaces. Operando Ambient Pressure X-ray photoelectron spectroscopy investigations provided quantitative understanding of the energy bands the the parameters what make these photoelectrochemical conduction.

X-ray techniques play an important role for gaining the fundamental understanding needed to tailor novel catalysts for CO2 reduction reaction (CO2RR), by providing chemical and structural information of catalytic surfaces. We have utilized surface-sensitive soft X-ray techniques to investigate the interaction of metal catalytic surfaces with electrolytes and/or gases (H2O and/or CO2) under in situ/operando conditions at the Advanced Light Source (ALS). This poster reports our work on AP-XPS for studying (i) CO2 adsorption on Cu and Ag surfaces to understand the initial atomic level events for CO2 electroreduction on the metal catalysts, (ii) CO2 adsorption on Ag/Cu alloys to provide the basis for developing improved catalysts electrolyte/solid catalytic surfaces.
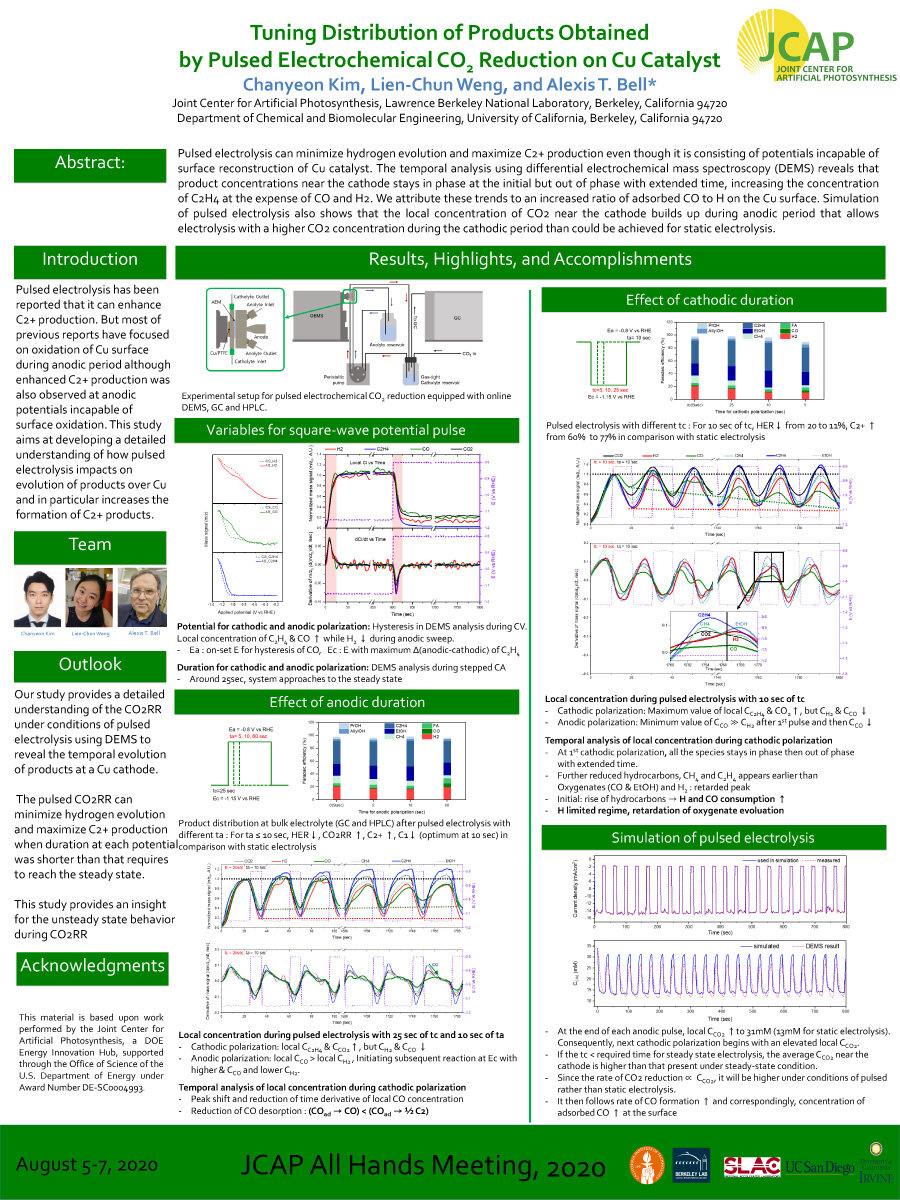
Pulsed electrolysis can minimize hydrogen evolution and maximize C2+ production even though it is consisting of potentials incapable of surface reconstruction of Cu catalyst. The temporal analysis using differential electrochemical mass spectroscopy (DEMS) reveals that product concentrations near the cathode stays in phase at the initial but out of phase with extended time, increasing the concentration of C2H4 at the expense of CO and H2. We attribute these trends to an increased ratio of adsorbed CO to H on the Cu surface. Simulation of pulsed electrolysis also shows that the local concentration of CO2 near the cathode builds up during anodic period that allows electrolysis with a higherCO2 concentration during the cathodic period than could be achieved for static electrolysis.
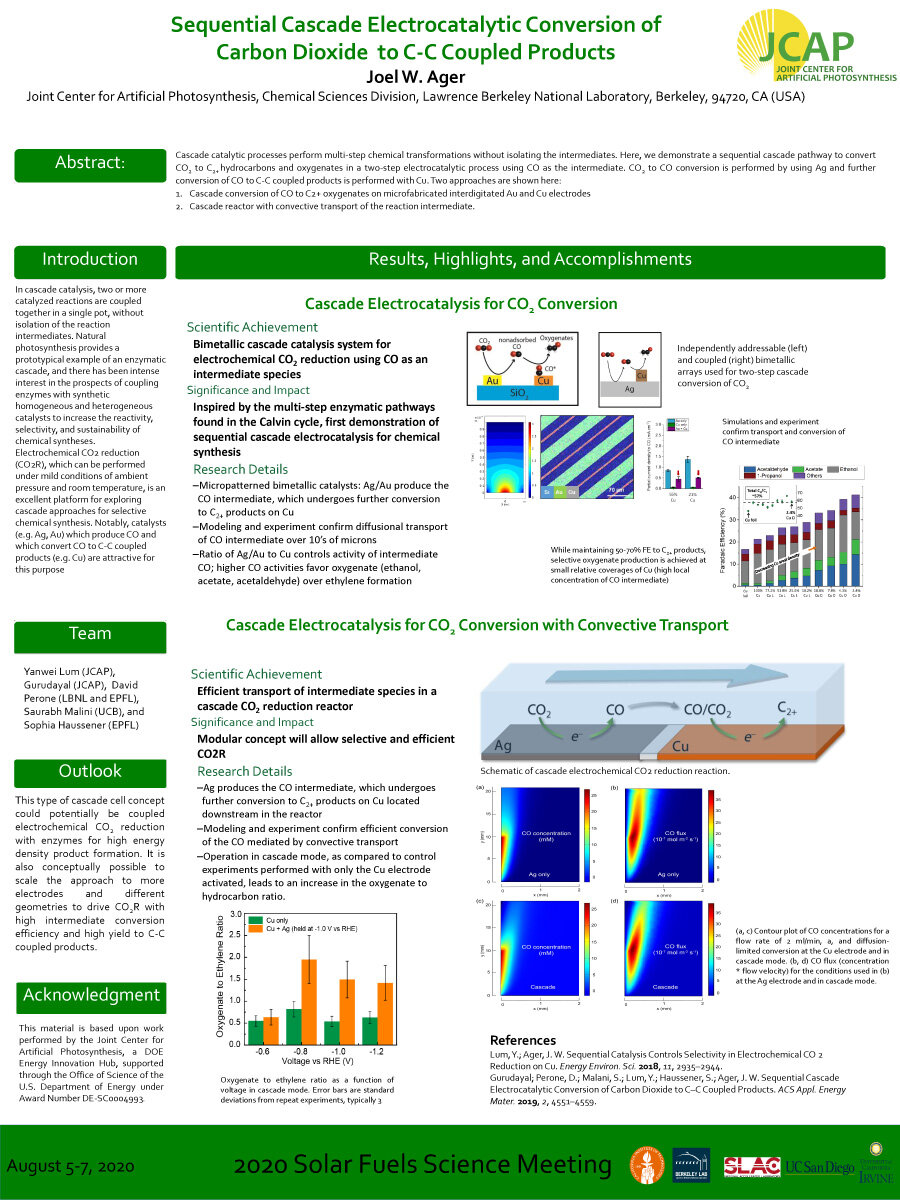
Cascade catalytic processes perform multi-step chemical transformations without isolating the intermediates. Here, we demonstrate a sequential cascade pathway to convert CO2 to C2+ hydrocarbons and oxygenates in a two-step electrocatalytic process using CO as the intermediate. CO2 to CO conversion is performed by using Ag and further conversion ofCO to C-C coupled products is performed with Cu.Two approaches are shown here: (a) Cascade conversion ofCO to C2+ oxygenates on microfabricated interdigitated Au and Cu electrodes; (b) Cascade reactor with convective transport of the reaction intermediate.

Due to the rapid depletion of fossil fuels and related environmental issues, developing technologies for producing renewable, clean fuels for our future is of
great importance. Artificial photosynthesis via water splitting or CO2 reduction offers an attractive and cost-effective route to achieve this goal. However,
existing (photo)electrocatalytic materials generally suffer from low activity or instability, thus greatly impeding the feasibility of artificial photosynthesis
technologies. Herein, we present design strategies for the synthesis and integration of novel (photo)electrocatalytic materials, which sheds light on rational design of photoelectrocatalytic materials for artificial photosynthesis.
Please contact us at jcapsciencemeeting2020@caltech.edu, if you have questions or would like to contact authors.
Digdaya et al.
In this poster, we demonstrate the proof-of concept system that can provide a unique technological pathway for CO2 capture and conversion using electrochemical processes.